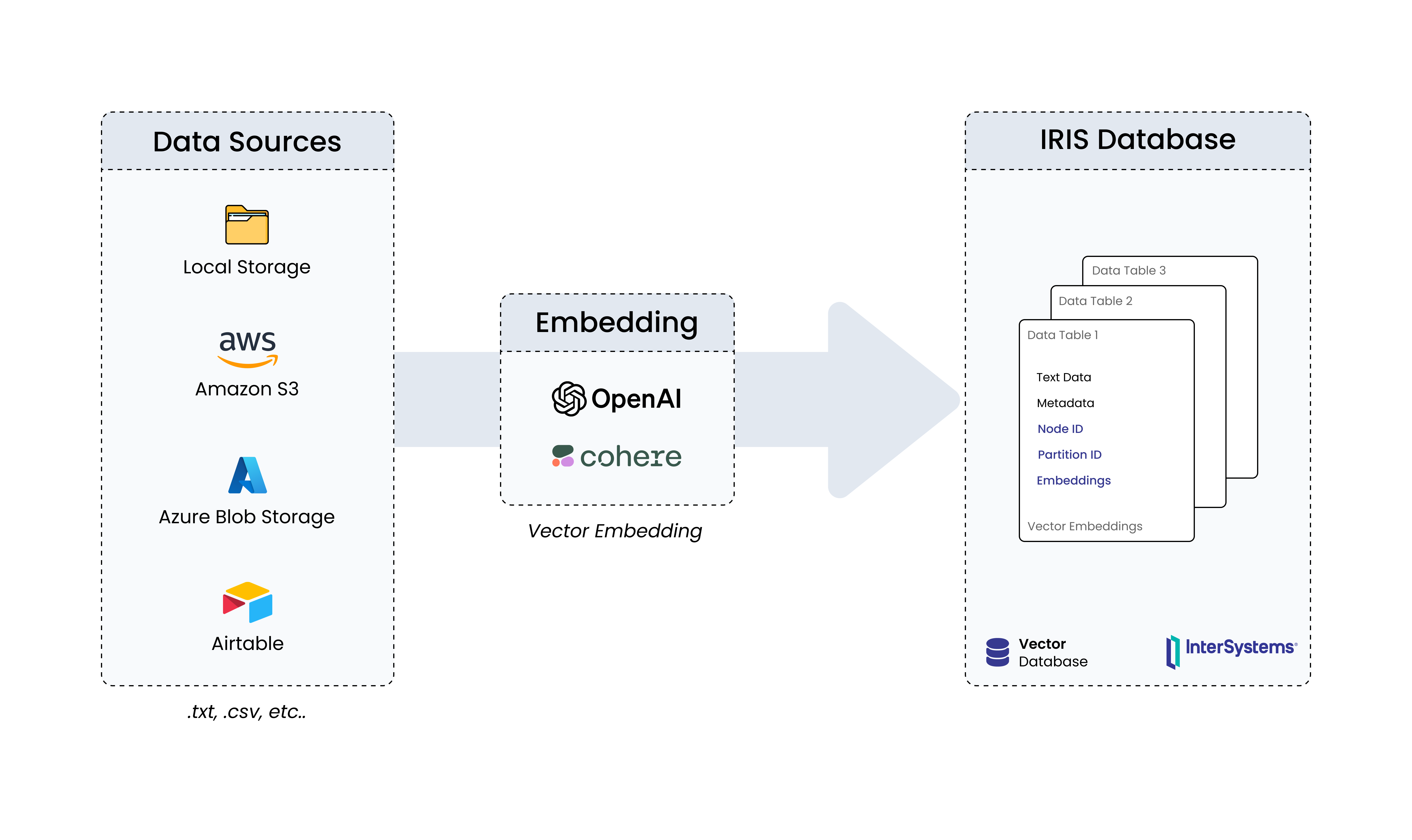
DNA Similarity and Classification was developed as a REST API utilizing InterSystems Vector Search technology to investigate genetic similarities and efficiently classify DNA sequences. This is an application that utilizes artificial intelligence techniques, such as machine learning, enhanced by vector search capabilities, to classify genetic families and identify known similar DNAs from an unknown input DNA.
.png)
K-mer Analysis: Fundamentals in DNA Sequence Analysis
Fragmentation of a DNA sequence into k-mers is a fundamental technique in genetic data processing. This approach involves breaking down the DNA sequence into smaller subsequences of fixed size, known as k-mers. For example, if we take the sequence "ATCGTAGCTA" and define k as 3, we would obtain the following k-mers: "ATC", "TCG", "CGT", "GTA", "TAG", "AGC", "GCT", and "CTA".
This process is carried out by sliding a window of size k along the sequence, extracting each subsequence of size k, and recording it as a k-mer. The window then moves to the next set of bases, allowing overlap between the k-mers. This overlap is crucial to ensure that no information is lost during the fragmentation process.
.png)
The resulting k-mers retain crucial local information about the structure of the DNA sequence. This is critical because many biologically relevant pieces of information are contained within specific regions of DNA. Subsequent analysis of these k-mers can reveal patterns, identify conserved or similar regions between different DNA sequences, and assist in the investigation of genetic functions and interactions.
In summary, DNA Similarity and Classification offer an approach to identify genetic similarities and classify DNA sequences.
Application
The following code initializes the project, performing the necessary setup before building:
ClassMethod init()
{
write "START INIT"
Do ##class(dc.data.DNAClassification).CreateTheModelML()
Do ##class(dc.data.HumanDNA).UploadData()
}
The UploadData method implemented in the code is responsible for fragmenting DNA sequences into k-mers, converting these k-mers into vectors using a specific encoding model, and then storing these vectors in the database.
print("Reading dataset")
human_df = pd.read_table('/opt/irisbuild/data/human_data.txt')
human_df = human_df.sample(n=1500, random_state=42)
To enhance performance and reduce processing time during the project's build phase, a sampling process is implemented, limiting the read to the first 1,500 records of the file. While this strategy speeds up the process, it's worth noting that a comprehensive analysis of the file could provide a more thorough understanding of the data. However, due to the commitment to efficiency and the pace of construction, this approach was adopted.
def getKmers(sequence, size=6, max_length=5000):
kmers = [sequence[x:x+size].lower() for x in range(len(sequence) - size + 1)]
kmers = [kmer for kmer in kmers if len(kmer) > 0]
if len(kmers) == 0:
return [sequence]
return kmers[:max_length]
The MAX_LEN parameter in getKmers is used to limit the maximum size of the k-mers processed to optimize the runtime of the process. However, it's important to note that this approach may not be ideal in terms of fully representing the DNA data.
print("Creating K-mers groups")
human_df['K_mers'] = human_df['sequence'].apply(getKmers)
print("Combining K-mers into strings")
human_df['K_mers_str'] = human_df['K_mers'].apply(lambda x: ' '.join(x))
print("Download stsb-roberta-base-v2 model")
model = SentenceTransformer('stsb-roberta-base-v2')
print("Encode K_mers")
embeddings = model.encode(human_df['K_mers_str'].tolist(), normalize_embeddings=True)
print("Creating column sequence_vectorized")
human_df['sequence_vectorized'] = embeddings.tolist()
The code vectorizes DNA sequences using the encoding model stsb-roberta-base-v2 from the SentenceTransformer library. This step converts DNA sequences into numerical vectors, facilitating their vector manipulation and computational analysis. After vectorization, the data is stored in the database for future use. This step is crucial to ensure that DNA sequence vectors are available for queries and additional analyses.
Subsequently, the method findSimilarity is responsible for finding the most similar DNA sequences based on a received DNA sequence input. It fragments the DNA sequence into k-mers and queries the database to find the five closest DNA sequences.
ClassMethod findSimilarity(pSequence As %String) As %String [ Language = python ]
{
import iris
from sentence_transformers import SentenceTransformer
results = []
def getKmers(sequence, size=6, max_length=5000):
kmers = [sequence[x:x+size].lower() for x in range(len(sequence) - size + 1)]
kmers = [kmer for kmer in kmers if len(kmer) > 0]
if len(kmers) == 0:
return [sequence]
return kmers[:max_length]
model = SentenceTransformer('stsb-roberta-base-v2')
kmers = getKmers(pSequence)
kmers_str = ' '.join(kmers)
search_vector = model.encode(kmers_str, normalize_embeddings=True).tolist()
stmt = iris.sql.prepare("SELECT TOP 5 ID FROM dc_data.HumanDNA ORDER BY VECTOR_DOT_PRODUCT(kMersVector, TO_VECTOR(?)) DESC ")
rs = stmt.execute(str(search_vector))
class_mapping = {
0: 'G protein coupled receptors',
1: 'tyrosine kinase',
2: 'tyrosine phosphatase',
3: 'synthetase',
4: 'synthase',
5: 'lon channel',
6: 'transcription factor',
}
for idx, row in enumerate(rs):
humanDNA = iris.cls("dc.data.HumanDNA")._OpenId(row[0])
results.append({
"sequence": humanDNA.sequence,
"dnaClass": class_mapping[humanDNA.dnaClass]
})
return results
}
These processes are essential for the project as they enable the comparative analysis of DNA sequences and the identification of patterns or similarities among them.
Applied Machine Learning
Machine learning has been utilized to perform DNA sequence classification. The DNAClassification class contains methods for training a multinomial Naive Bayes classification model and for classifying DNA sequences based on this model.
The CreateTheModelML method is responsible for training the classification model. Firstly, DNA sequences are preprocessed, transformed into k-mers, and grouped. Then, these k-mer groups are converted into a numerical representation using vectorization technique. The multinomial Naive Bayes model is then trained with these vectorized data. After training, the model's performance is evaluated through analyses such as precision, recall, and F1 score.
ClassMethod CreateTheModelML()
{
import pandas as pd
def getKmers(sequence, size=6):
kmers = [sequence[x:x+size].lower() for x in range(len(sequence) - size + 1)]
kmers = [kmer for kmer in kmers if len(kmer) > 0]
if len(kmers) == 0:
return [sequence]
return kmers
print("Reading dataset")
human_df = pd.read_table('/opt/irisbuild/data/human_data.txt')
print("Creating K-mers groups")
human_df['K_mers'] = human_df['sequence'].apply(getKmers)
human_df['words'] = human_df.apply(lambda x: getKmers(x['sequence']), axis=1)
human_df = human_df.drop('sequence', axis=1)
human_texts = list(human_df['words'])
for item in range(len(human_texts)):
human_texts[item] = ' '.join(human_texts[item])
y_data = human_df.iloc[:, 0].values
from sklearn.feature_extraction.text import CountVectorizer
cv = CountVectorizer(ngram_range=(4,4))
X_human_dna = cv.fit_transform(human_texts)
print(X_human_dna.shape)
print("PREPARING FIT DATA")
from sklearn.model_selection import train_test_split
X_train, X_test, y_train, y_test = train_test_split(X_human_dna,
y_data,
test_size = 0.20,
random_state=42)
print("FIT THE MODEL")
from sklearn.naive_bayes import MultinomialNB
classifier = MultinomialNB(alpha=0.1)
classifier.fit(X_train, y_train)
y_pred = classifier.predict(X_test)
print("VALIDATING THE MODEL")
from sklearn.metrics import accuracy_score, f1_score, precision_score, recall_score
print("Confusion matrix\n")
print(pd.crosstab(pd.Series(y_test, name='Actual'), pd.Series(y_pred, name='Predicted')))
def get_metrics(y_test, y_predicted):
accuracy = accuracy_score(y_test, y_predicted)
precision = precision_score(y_test, y_predicted, average='weighted')
recall = recall_score(y_test, y_predicted, average='weighted')
f1 = f1_score(y_test, y_predicted, average='weighted')
return accuracy, precision, recall, f1
accuracy, precision, recall, f1 = get_metrics(y_test, y_pred)
print("accuracy = %.3f \nprecision = %.3f \nrecall = %.3f \nf1 = %.3f" % (accuracy, precision, recall, f1))
print("SAVE THE MODEL")
import joblib
joblib.dump(classifier, '/opt/irisbuild/data/multinomial_nb_model.pkl')
joblib.dump(cv, '/opt/irisbuild/data/cv_to_multinomial_nb_model.pkl')
}
On the other hand, the ClassifyDnaSequence method is responsible for classifying the received DNA sequences as input. Firstly, the sequence is preprocessed and transformed into k-mers. Then, these k-mers are vectorized using the same process used during the model training. The trained model is then loaded and used to predict the class of the DNA sequence. The classification result includes the predicted class and the probabilities associated with each class.
ClassMethod ClassifyDnaSequence(sequence As %String) As %Status
{
import joblib
from sklearn.feature_extraction.text import CountVectorizer
classifier = joblib.load('/opt/irisbuild/data/multinomial_nb_model.pkl')
cv = joblib.load('/opt/irisbuild/data/cv_to_multinomial_nb_model.pkl')
def getKmers(sequence, size=6):
kmers = [sequence[x:x+size].lower() for x in range(len(sequence) - size + 1)]
kmers = [kmer for kmer in kmers if len(kmer) > 0]
if len(kmers) == 0:
return [sequence]
return kmers
k_mers = getKmers(sequence)
k_mers_vec = cv.transform([' '.join(k_mers)])
predicted_class = classifier.predict(k_mers_vec)[0]
probabilities = classifier.predict_proba(k_mers_vec)[0]
class_mapping = {
0: 'G protein coupled receptors',
1: 'tyrosine kinase',
2: 'tyrosine phosphatase',
3: 'synthetase',
4: 'synthase',
5: 'lon channel',
6: 'transcription factor',
}
result = {
"Classification": class_mapping[predicted_class],
"Probabilities": {
class_mapping[class_index]: float(probability)
for class_index, probability in enumerate(probabilities)
}
}
return result
}
Usage of the Application
Users interact with the system by sending DNA sequences through queries and can request a JSON-formatted response containing information about the discovered similar sequences and their classifications, simplifying the process of genetic analysis.
Below is the API to be used: http://localhost:52773/api/dna/find?&dna=<YOUR_DNA>
- dna: Enter the DNA sequence you want to search
Example: http://localhost:52773/api/dna/find?&dna=ATGAACTGTCCAGCCCCTGTGGAGATCTCCT...
Using Postman to visualize the API response (can be done using a browser or other applications like Insomnia):
.png)
Project: GitHub
Voting: DNA-similarity-and-classify
 Open Exchange
Open Exchange
.png)
.png)
.png)
.png)
.png)
.png)